Unravelling evolutionary pathways is key to understanding molecular mechanisms, providing insights into how genes and proteins have evolved to acquire their current functions. However, molecular biologists sometimes appear indifferent to evolution, questioning its relevance to mechanistic research.
In recent years, my lab — or perhaps it’s just me — has developed an obsession with evolutionary transitions. The view that every gene originates from an ancestral state and undergoes impactful changes through its evolutionary journey, whether it’s the gain or loss of an activity or function. The challenge lies in meticulously mapping out these key evolutionary innovations that have significantly influenced function. Addressing this challenge is not merely interesting but absolutely essential in biology. Our aim as biologists transcends understanding how biological systems operate; we seek to unravel how they came to be. And the two questions are more connected than many think.
This post stems from my observation that molecular biologists sometimes appear indifferent to evolution, questioning its relevance to mechanistic research. It baffles me why the centrality of evolution in biology isn’t apparent to some. Maybe they’ve never taken a course on the subject, or perhaps they’ve never fully appreciated the profound concept that every organism and every gene is connected through an unbroken chain of descent to countless ancestors. This perspective holds profound implications for mechanistic molecular biology.
If you already appreciate the link between evolutionary biology and molecular mechanisms, you might find this post to be music to your ears. However, if you’re among those who question the value of evolutionary biology, I encourage you to stay with me; you might discover its significance in ways you hadn’t considered before.

What are evolutionary transitions?
In the context of this article, “evolutionary transitions” is the straightforward way to describe what evolutionary biologists call a derived trait — a new trait or mutation that has emerged over time from a distinct ancestral state. As Sean B. Carroll eloquently puts it in his engaging book “The Making of the Fittest”, evolutionary transitions are about “making the new out of the old”.
In evolutionary biology, particularly when focusing on organisms, there’s a strong emphasis on major evolutionary transitions. Take, for instance, the evolution of flowering plants (angiosperms). The presence of flowers represents a derived trait in plants that originated in a single common ancestor, diverging from other non-flowering ancestors.

In the context of molecular evolution, evolutionary transitions can be as straightforward as a single nucleotide change within an organism genome that confers a new characteristic to a gene, diverging from its ancestral sequence.
An example of this comes from research conducted by Ryohei Terauchi at Kyoto University and Iwate Biotechnology Research Institute, Mark Banfield at John Innes Centre and colleagues on a virulence effector family in the plant pathogenic blast fungus, Magnaporthe oryzae. Specifically, genes within the AVR-Pik family exhibit multiple non-synonymous substitutions. These are the nucleotide polymorphisms that result in a change in the encoded amino acid within the effector protein, potentially modifying the protein function and leading to adaptive evolution (see “The one thing you need to know about molecular evolution” for more insights on this subject).
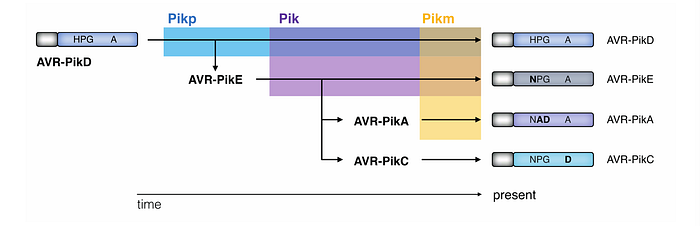
In this context, a derived trait can consist of a single nucleotide polymorphism. For example, as illustrated below, the highlighted C to A mutation in position 46 of the AVR-Pik gene coding sequence has modified the codon from the ancestral CAC to the derived AAC, therefore altering the protein sequence from Histidine (H) to Asparagine (N). Whereas AVR-PikA, F, E and C alleles carry the derived sequence (AAC), AVR-PikD as well as all the variants of the related paralogous (duplicate) genes APikL1 and APikL2 carry the ancestral sequence of CAC.
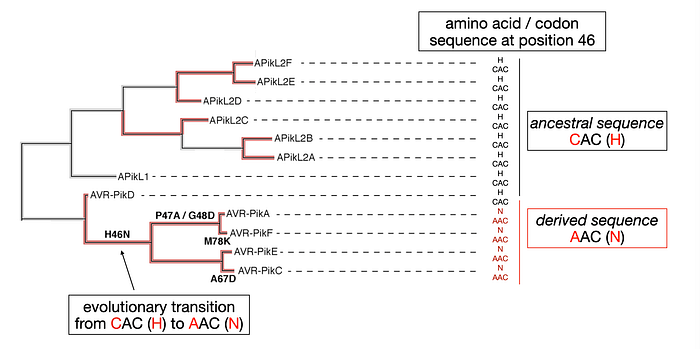
In this example the evolutionary transition from CAC (H) to AAC (N) has emerged during AVR-Pik gene evolution on the branch indicated with the arrow.
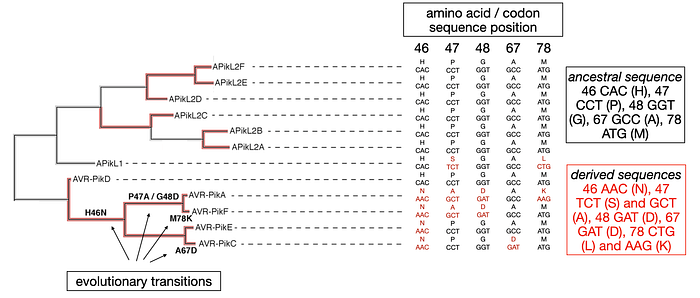
The evolution of AVR-Pik involved more than just the previously mentioned transition. As depicted in the figure below, four additional non-synonymous substitutions appeared during the evolution of the AVR-Pik alleles at positions 47, 48, 67, and 78. These substitutions occurred in the branches leading to the AVR-PikA/F, E, and C alleles, marking them as derived sequences, with AVR-PikD maintaining the ancestral codon sequence in each instance. It’s also worth noting that APikL1, a related gene, features two derived codon sequences at positions 47 and 78 (changing from CCT to TCT and ATG to CTG, respectively), further illustrating the complex nature of evolutionary changes within this effector gene family.
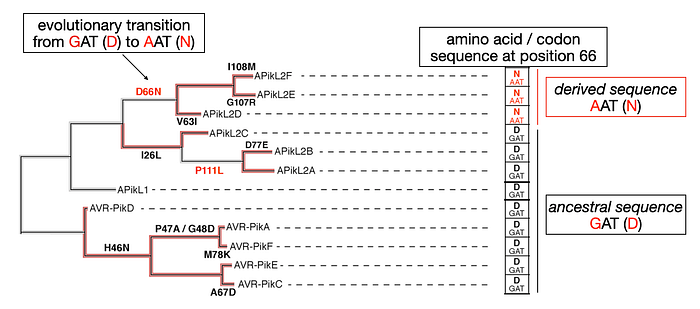
Thorsten Langner and colleagues have investigated the evolution of the APikL2 gene. This gene mirrors the ancestral state of AVR-PikD at five specific positions mentioned earlier. Nonetheless, APikL2 exhibits a unique array of derived mutations. For instance, a mutation from G to A at position 66 in the APikL2 gene coding sequence changes the codon from the ancestral GAT to the derived AAT, thus altering the protein sequence from Aspartate (D) to Asparagine (N). This derived sequence (AAT) is present in APikL2D, E, and F, whereas APikL2A, B, and C alleles, along with APikL1 and all AVR-Pik alleles, maintain the ancestral GAT sequence.
This particular evolutionary transition from GAT (D) to AAT (N) as well as other non-synonymous mutations in the APikL2 gene are highlighted in the figure, showcasing the distinct evolutionary pathways that have shaped this gene over time.
The value of ancestral sequence reconstructions
How did we ascertain that, in the example mentioned above, AAT (N) represents the derived sequence and GAT (D) the ancestral one? Credit goes to University of Chicago evolutionary biologist Joe Thornton, who pioneered the method known as ancestral sequence reconstruction (ASR).
Ancestral sequence reconstruction (ASR) is used to infer the genetic sequence of ancient organisms by analyzing the sequences of their contemporary descendants. Through comparative genomics and phylogenetic analysis, ASR allows scientists to estimate the sequences of common ancestors at various nodes in the evolutionary tree. This method involves creating a phylogenetic tree to represent the evolutionary relationships among a group of genes or species. By applying statistical models to this tree, researchers can work backward from the known sequences of modern organisms to predict the most likely sequences of their ancestors.
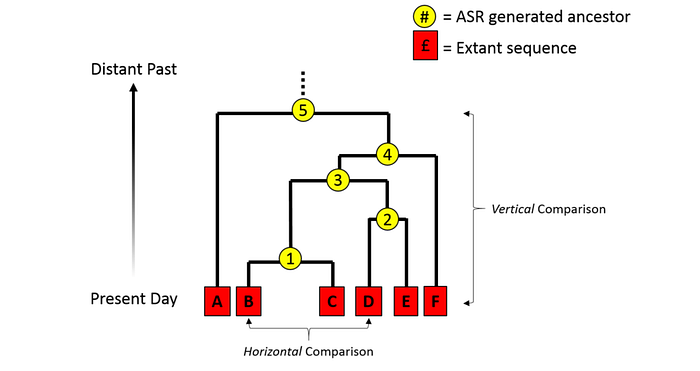
Consequently, ASR provides us with the tools to accurately trace molecular evolutionary transitions, establishing directionality by identifying which sequences are ancestral and which are derived. Therefore, whenever you are comparing related gene or protein sequences, ask yourself which is ancestral and which is derived. This knowledge will help you frame experiments and interpret biochemical and functional data.
Unravelling evolutionary pathways informs molecular mechanisms
Ancestral reconstructions at pivotal phylogenetic nodes illuminate the evolutionary trajectories of genes and proteins, revealing transitions and adaptive features that subsequently inform molecular mechanisms. By distinguishing between ancestral and derived sequences, we acquire crucial insights, enabling us to place functional molecular changes into a broader mechanistic framework.
Lets examine the examples above to understand how ancestral reconstructions and unravelling evolutionary pathways can inform mechanistic models.
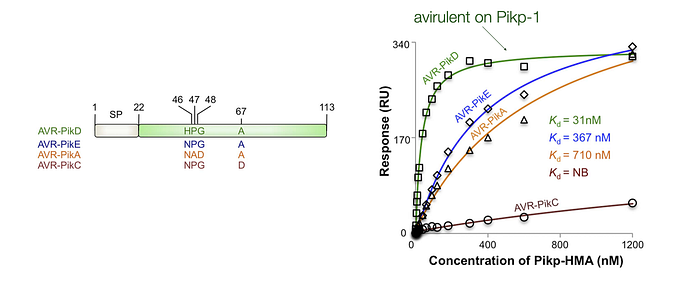
Taking the AVR-Pik example, we established that AVR-PikD aligns with the ancestral state and identified five derived mutations in AVR-PikA, F, E, and C at positions 46, 47, 48, 67, and 78. These mutations have emerged within the fungal population to circumvent activation of the rice immune receptor, Pikp. Consequently, blast fungus strains carrying the AVR-PikA, F, E, and C alleles can infect rice plants possessing the Pikp receptor, unlike those with AVR-PikD. In this work, a convergence of genetic, biochemical and evolutionary studies is what enabled us to develop this model.
It turned out that the rice immune receptor Pikp-1 directly binds the effector protein AVR-PikD. Abbas Maqbool and colleagues employed biophysical techniques to assess how the derived mutations affect the binding affinity. Their findings indicate that AVR-PikE, A, and C exhibit reduced binding affinity to Pikp-1 compared to AVR-PikD. This suggests that the mutations in these AVR-Pik alleles have allowed the pathogen to elude detection by this immune receptor.
Further, the mutations of the four amino acids at positions 46, 47, 48, and 67 in AVR-PikE, A, and C are located directly at the binding interface between the Pikp-1 receptor and the pathogen effector. This precise localization explains the impact of these mutations on binding affinity, highlighting their role in the pathogen ability to avoid immune detection through derived mutations that alter the binding dynamics between the receptor and effector.
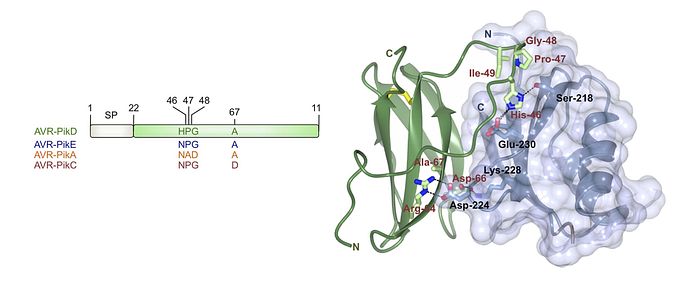
This example underscores the significance of ancestral reconstructions and the unravelling of evolutionary pathways in informing and refining our mechanistic models. The integration of genetic, biochemical, and evolutionary studies is what enabled the development of our model, elucidating how derived mutations in the AVR-PikE, A, and C originated from the ancestral AVR-PikD effector to circumvent detection by the disease resistance receptor Pikp and other alleles of this receptor. Without the evolutionary perspective, we would struggle to interpret the emergence and establishment of these polymorphisms within the pathogen population.
Ancestral reconstructions have also facilitated the development of a plausible model for the coevolution between the APikL2 effector of the blast fungus pathogen and its host grass plants. As previously mentioned, these reconstructions have clarified the directionality of evolution, revealing that the ancestral variant of the APikL2 effector, D66 (GAT codon), preceded the derived form, N66 (AAT codon). Thorsten Langner and colleagues demonstrated that the D66N polymorphism broadens the ability of APikL2 to interact with host proteins from the sHMA family. Specifically, APikL2 variants harboring the derived Asparagine (N) at position 66 are capable of binding sHMA94 from the grass plant Setaria italica, unlike their ancestral counterparts with Aspartate (D) at position 66. This insight suggests that the D66N polymorphism enabled an expansion in host-target binding capabilities rather than evasion of host immunity. The model is, therefore, quite different from the evolution of AVR-Pik described above, with D66N functioning as an adaptive trait that enhances binding to novel host targets, potentially contributing to adaptation to different host plants.
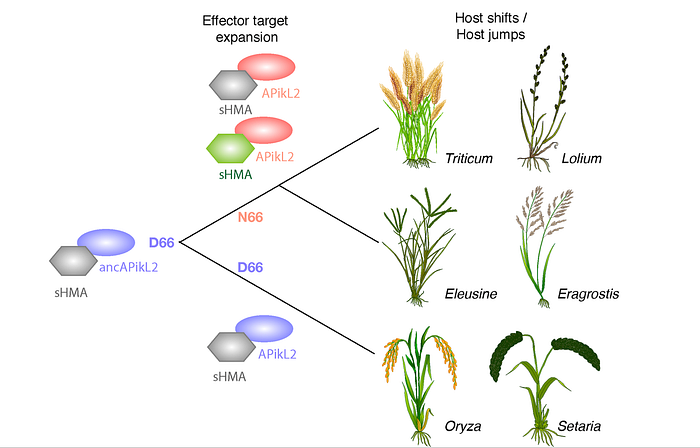
Hence, ancestral reconstructions of the related effectors AVR-Pik and APikL2 have enabled the formulation of contrasting mechanistic models explaining the emergence of derived mutations in these effectors and their functional impacts within this pathosystem. Without the evolutionary perspective, distinguishing between these mechanistic models would have been particularly challenging.
Unravelling evolutionary pathways informs experimental design
Reconstructing evolutionary pathways can be crucial for informing experimental design, with ancestral reconstructions playing a pivotal role in shaping mechanistic experiments.
Let me use a rather original example to illustrate this point. Consider the study of bird wings, comparing flightless birds like ostriches to their flighted counterparts. Understanding that the wings of ostriches are a derived trait from those used for flight aids in comprehending the physiological and molecular adaptations that have led to their current functions.
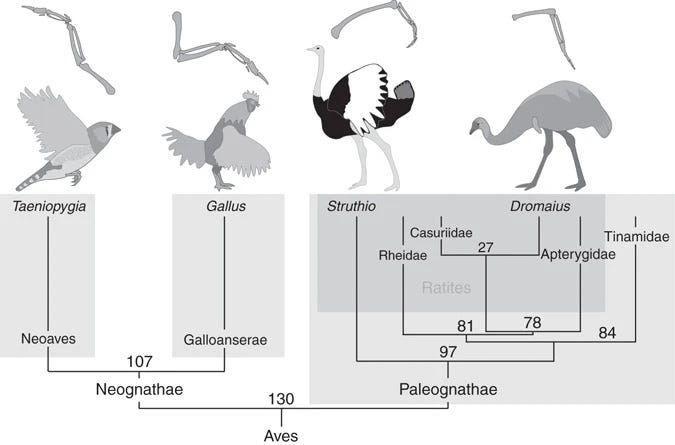
Recognizing evolutionary transitions is key to designing appropriate experiments. In the context of ostriches, the evolutionary framework suggests that gain-of-function experiments — such as grafting wings from a flighted bird onto an ostrich — would likely yield inconclusive results. This is because, through evolution, ostriches have undergone numerous changes related to their flightless state. Flightlessness is what we would call a highly derived trait.
In contrast, for this scenario, the evolutionary model would point to grafting ostrich wings or even a knock-out loss-of-function experiments in a flighted bird as being more likely to yield valuable insights. Similarly, when considering mutations in a gene or protein based on comparative analyses, understanding the evolutionary history is imperative for devising an optimal experiment. For example, a protein might be as biochemically derived for its activity as an ostrich is in terms of flightlessness. To figure this out, ancestral sequence reconstructions are necessary to develop an evolutionary model that guides your experimental approach.
Don’t neglect regressive evolutionary transitions
As illustrated by the evolution of bird wings, evolutionary transitions do not solely have to be functional innovations but can also encompass loss of function. Such transitions include gene deletion, pseudogenization all the way to the disappearance of specific biochemical features, all of which contribute valuable insights to mechanistic models. This underpins my preference for the term “evolutionary transition” over the more commonly employed “evolutionary innovation.” Indeed, there is a tendency in evolutionary biology to focus on the acquisition of new functions (neo-functionalization), overlooking the prevalence and significance of sub-functionalization and loss of function. These instances represent regressive evolutionary transitions, which do not align neatly with the notion of “evolutionary innovations.”
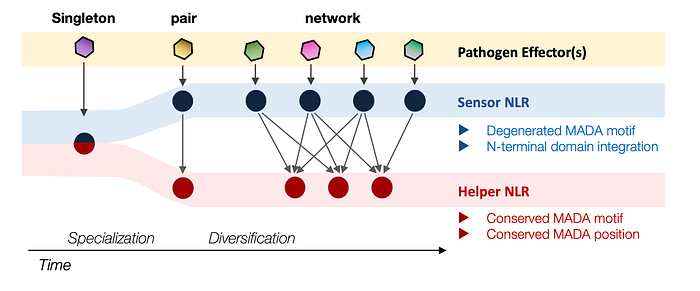
The importance of recognizing regressive evolution is exemplified in the study of plant immune receptors of the NLR class, where some receptors have diverged from an ancestral, multifunctional receptor capable of both detecting pathogen effectors and executing immune responses into specialized (sub-functionalized) classes of sensors and helpers. This specialization often entails the loss of specific biochemical features. For instance, the degeneration of the N-terminal MADA motif in a subset of sensor NLRs through evolution exemplifies such a loss. Reconstructing the evolutionary trajectory from this ancestral N-terminal motif, crucial for initiating the immune response, to a non-functional one is vital for a comprehensive mechanistic understanding of these sensor receptors. Without this evolutionary perspective, molecular biologists might waste considerable effort pursuing non-existent activities and designing flawed experiments.
The opportunity: a genome data deluge
Why the sudden interest in molecular evolution? This burgeoning interest in evolutionary studies, despite seeming familiar to many, is primarily fuelled by the exponential growth in available genome data. In recent years, the accumulation of extensive genome datasets has enabled meaningful comparative analyses that were previously unfeasible. Consider the example of plant genomes: historically, assembling these genomes to chromosome scale was both challenging and costly. Yet, as of now, we have hundreds of high quality plant genomes, with new additions from a broad spectrum of plant diversity emerging almost daily.
For instance, my Sainsbury Lab colleagues Yu Sugihara, AmirAli Toghani and Jiorgos Kourelis have compiled a comprehensive dataset of 66,665 NLR sequences solely from 124 plant species in the Solanaceae family. This compilation is invaluable, and has already facilitated evolutionary reconstructions of the potato immune receptor PERU and revealed that the homodimerization interface of NLR helper receptors within the NRC family has diversified over time.
The deluge of genome data, a continuous influx of high-quality plant and microbial genomes, underscores the relevance of adopting an evolutionary perspective in our current research. Furthermore, the recent advancements in protein structural biology, propelled in part by AI-driven tools like AlphaFold2, offer a unique opportunity to integrate multiple research approaches. This convergence aims to bridge the gap between evolutionary biology and mechanistic studies, leveraging the wealth of genome data to enhance our understanding of biological processes.
To sum up. I just have three words for you. Think. Evolutionary. Transitions.

Acknowledgements
I’m grateful to the many colleagues who inspired this article. This post was written with assistance from ChatGPT.
This article is available on a CC-BY license via Zenodo. Cite as: Kamoun, S. (2024) Think. Evolutionary. Transitions. Zenodo. https://doi.org/10.5281/zenodo.10653729
Additional resources and suggested readings
The one thing you need to know about molecular evolution

Evolution 101 — Stop teaching most mutations are neutral?

For more details on EVO-MPMI, check: Schornack, Sebastian and Kamoun, Sophien. (2023). EVO-MPMI: From fundamental science to practical applications. Zenodo https://doi.org/10.5281/zenodo.8030196
The ancient guardian: ZAR1 evolutionary journey and adaptations

Can a biologist fix a smartphone? — Just hack it!